Rather than relying on serendipity to discover novel therapeutics, today we can work intelligently to identify antibodies that target exactly what we desire. Our immune system has been refined through millions of years of evolution, and our ability to harvest its capabilities now allows us to develop intelligently designed antibody-based medicines. This new generation of biotherapeutics holds great promise in tackling diseases and conditions that were previously untreatable.
In the history of medicine, we have relied much on serendipity to find drugs that could alleviate diseases. However, in the late 18th century, a more targeted approach was beginning to see the light of day. Animals were immunized, their blood extracted, and the serum separated, in order to obtain their antibodies for so-called serotherapy. Before the discovery of antibiotics, this approach was used to treat bacterial infections. Today, our understanding of how serotherapy works has been the foundation of using antibodies as targeted therapies against diseases.
Antibodies are found in immune systems of all vertebrate animals. When infected with bacteria or viruses, B-cells (white blood cells) produce antibodies to fight the infection. These antibodies are excellent therapeutic molecules and are capable of recognizing exact targets of interest to neutralize (called antigens). Antibodies work by binding to and inhibiting antigens, thus preventing their potentially harmful effects, but can also be designed to activate an antigen. Nowadays antibody-based therapeutics are being used to treat diseases and conditions that, to date, have been difficult to combat by other means. These include cancer, autoimmune diseases, and donor-organ rejection.
To produce antiserum for serotherapy, antigens or pathogens (disease-causing organisms) are injected into production animals, such as horses or sheep. Antibodies from the serum of the production animal can then be extracted, yielding antiserum. However, as the antibodies in the antiserum are not of human origin, our immune system will see these antibodies as foreign and mount an attack. This can result in adverse reactions, which in rare cases can be lethal. To avoid these side effects, the antibodies we use as therapeutics today are typically either fully human or ‘humanized’. As an example, the drug Avastin (with the bioactive component/antibody, bevacizumab, that targets vascular endothelial growth factor VEGF) is humanized. This is done by replacing as many non-human parts of the antibody as possible with human antibody parts, without altering the specific regions that confer antigen binding. Therefore, the human immune systems allow Avastin to exert its therapeutic purpose with minimal risk of adverse reactions.
Although the term “antibody” is used, there are many types of antibodies, or immunoglobulins (Igs), to give them their scientific name. Globulins are globular proteins that flow in the circulatory system and are further divided into three classes (alpha, beta, and gamma globulins), with the gamma class containing the immunoglobulins. The most effective immunoglobulin that our body produces in response to pathogen infections have a gamma heavy chain (‘G’ type) and therefore are more commonly known as IgGs. These immunoglobulins are produced by the immune system’s B-cells, and each B-cell line makes its own, unique IgG.
IgG molecules consist of two types of protein chains, namely light and heavy chains, shown in Figure 1. On each chain, the outermost regions, variable regions (the “arms” on Figure 1), are responsible for antigen binding. The arms are also known as Fragment Antigen-Binding (Fab) and contain so-called Complementarity Determining Regions (CDRs), which contain hypervariable regions of amino acids. These hypervariable regions are where the IgGs differ from one another, and what confers their ability to recognize different antigens. There are three CDRs in each variable region, in both the light and heavy chain, yielding a total of 12 CDRs per IgG.
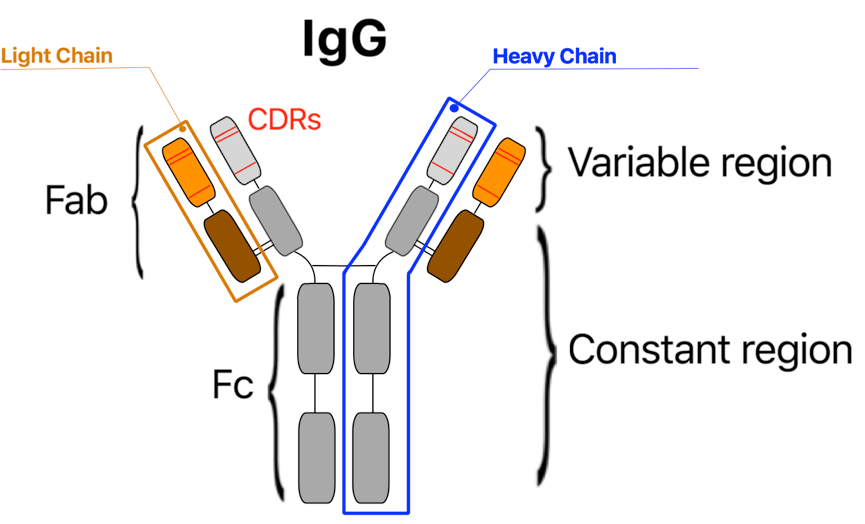
Figure 1: Schematic representation of the IgG antibody; its Fab fragments (brown & orange), Fc fragment, and heavy chains (grey), including the variable and constant regions. CDRs are marked in light grey. (Illustration: Albert Fuglsang-Madsen).
If an antibody-based medicine consists solely of identical IgGs (with identical variable regions), they all have identical antigen specificity and are collectively referred to as monoclonal antibodies (mAbs; e.g. Avastin – as mentioned above). In contrast, when isolating antibodies from immunized animals, what is obtained is a mix of antibodies with diverse antigen specificities due to non-identical CDRs. These mixes are referred to as polyclonal antibodies. However, it is not only the variable regions of antibodies that are important for their therapeutic properties; in the “tail” of the IgG antibody, the organism-specific Fragment crystallizable (Fc) region of the antibody is located. This region is very important for the antibody’s biological function, as it interacts with and activates the various immune cells. It also confers the long half-life of the antibody in our bloodstream.
INFOBOX1: The name of the immune system’s B-cells originates from “Bursa fabricii”, which is the organ in chickens from where they were originally isolated.
The Fc region is called “Fragment crystallizable” as it was the first part of the antibody that was crystallizable.
To be efficacious, our antibodies not only need impressive functional specificity but also great variety. Luckily, our immune system is capable of producing about 2.6 million (more when taking mutations into account) possible IgG combinations. Yet, although all B-cell lines produce their own unique IgG, not all of these possible combinations are encoded in our DNA; that would make our DNA huge! In fact, increasing the number of our protein-encoding genes from currently ~20,000 to 2.6 million would be equivalent to expanding an average human being to the size of the Eiffel tower!
Fortunately, our DNA is much more size-efficient. The different IgG components are encoded by genes that are cut and pasted into different combinations in B-cells as they mature, rather than having a copy of each combination. Shuffling the genes which encode the variable regions of IgG makes the resulting IgGs diverse and allows the CDRs to recognize different antigens. This is why IgGs are excellent therapeutic molecules. Whether you want to utilize IgGs to neutralize toxins, bacterial or viral infections, or inhibit tumor growth, these different combinations of the variable regions can be used to customize therapeutic antibody candidates, which can easily be tested in the lab.
The way the process of IgG maturation works is the following: When we are infected by pathogens, the shuffled sequences of the variable regions are tested against the invaders. If a B-cell reacts to one of the molecules of the pathogen, other immune cells will take notice and activate the corresponding B-cell. This activation initiates cell division to increase expression of specific antibodies and create memory cells that remember the invading antigen for a long period of time. The memory cells can then be reactivated, should the same pathogen ever be encountered again. By intentionally exposing humans to inactivated pathogens or antigenic molecules from pathogens, we can simulate an infection and have the immune system build up memory cells. If we are eventually infected by that pathogen, the memory cells recognize it and start producing the necessary antibodies right away. This is the principle behind vaccination.
How are antibody-based therapeutics produced?
Our immune system has evolved over millions of years specifically to defend our bodies from infection and it would be a tragedy not to take advantage of that. Therefore, many new technologies mimic or even utilize the immune system’s ability to produce antibodies against relevant antigens.
One example of this is hybridoma technology. Here, typically a mouse is immunized, meaning that it will be exposed to low doses of the relevant antigen over a period of time. The dose is typically gradually increased, causing antibodies against the antigen to be expressed by the B-cells. In the end, the mouse will have built up a strong immune response to the antigen (such as a toxin), and the B-cells can be isolated from the mouse’s blood or spleen. These cells are then fused with immortal cancer cells (i.e. myeloma cells), which can be maintained in a Petri dish in the lab or cultured in bioreactors for large-scale production. One example of this is hybridoma technology (see Figure 2).
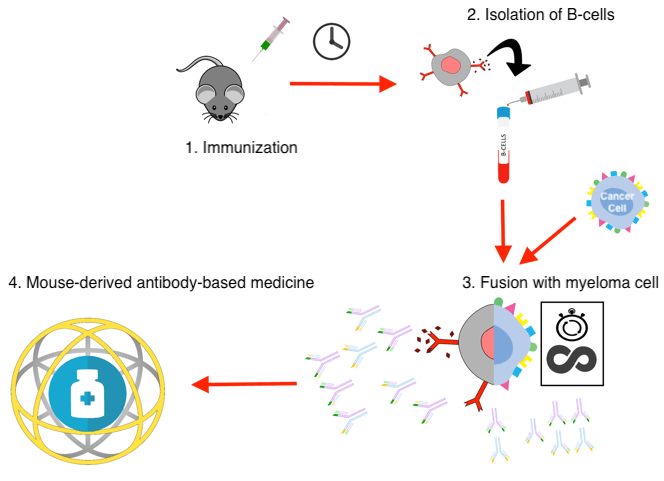
Figure 2: Schematic overview of the hybridoma technology. (1) A mouse is immunized with the desired antigen. The B-cells are subsequently isolated (2) and fused with cancer (myeloma) cells (3), which can then mass-produce the mouse-derived antibodies indefinitely (4). (Illustration: Albert Fuglsang-Madsen).
One of the challenges of hybridoma technology is that it is laborious and can take a long time to build up the immunity of the animal to the desired antigen. Furthermore, once the animal’s B-cells are isolated, screening approaches need to be used to determine which B-cell expresses the desired antibody that is specific against the antigen. Also, the Fc part of the antibody is specific to each animal, which means that the human immune system will recognize it as foreign, which can result in adverse reactions, as mentioned previously.
There have been recent advances in hybridoma technology, such as transgenic mice (and other animals). Transgenic mice have human variable regions encoded in place of the variable regions of their own mouse antibodies. When the genetic sequences for such transgenic animal-derived antibodies are obtained, the antibodies can be re-engineered by combining the human variable regions, obtained from the transgenic mouse, with the human Fc region to create a fully human IgG (see Figure 3). Such a fully human IgG can be used for human therapy with far fewer adverse reactions and even with increased efficacy. However, immunization and screening still require significant resources, and transgenic animals remain costly to procure or develop. Nonetheless, the technology is already used by companies such as Symphogen, Kymab, and Trianni, who have developed effective antibodies to treat different cancers, autoimmune disorders, and rejection of transplanted organs.
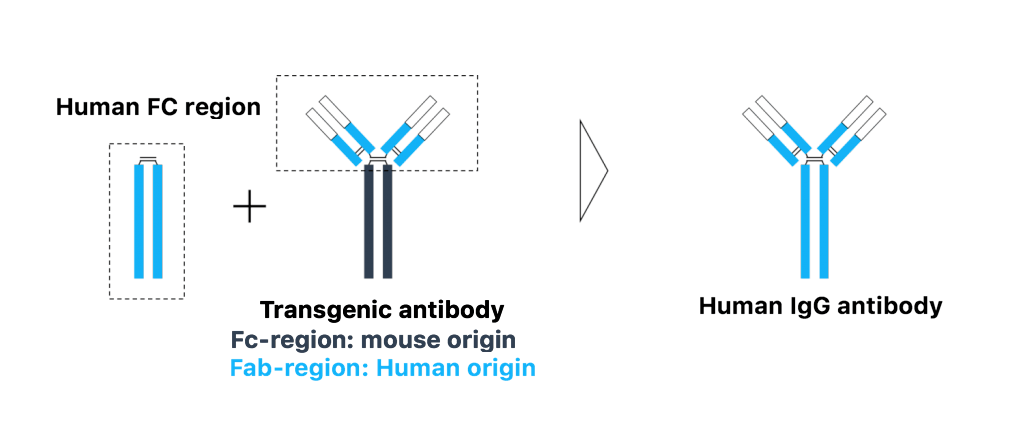
Figure 3: By taking the variable regions (human) of a transgenic antibody, and combining them with a human Fc region, it is possible to create a 100% human antibody, which is compatible with the human immune system. (Illustration: Andreas Hougaard Laustsen).
There are also other useful technologies in addition to hybridoma technology to discover antibodies with specificity to the desired antigen. One of these is phage display technology, a method invented by George P. Smith in 1985, in which a short sequence of amino acids can be expressed and displayed on the surface of bacteriophages (bacterial viruses). Five years after Smith’s original invention, John McCafferty successfully used the method to display human antibody fragments, specifically the antigen binding regions (see Figure 4).
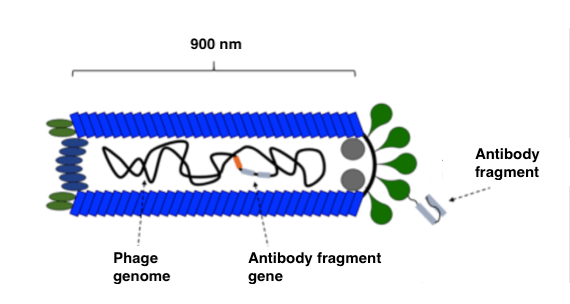
Figure 4: Schematic representation of a bacteriophage. The green and blue shapes represent proteins, which comprise the bacteriophage’s coat. In the far right, the antibody fragment is displayed, attached to a green coat protein. The corresponding gene for the antibody fragment is found inside the bacteriophage in its genome. (Illustration: Andreas Hougaard Laustsen).
Phage display experiments consist of a four-step process (see Figure 5). 1) Panning a diverse collection of phages, carrying antibody fragments, into a vial in which relevant antigens have been immobilized; 2) washing away the phages that do not bind the antigens; 3) releasing the bound phages from the immobilized antigens, and 4) infecting E. coli bacteria with the bound phages to amplify the number of each phage. After the amplification step, one or more rounds of panning may be performed to create further competition between weakly-binding and strongly-binding phages (see Figure 5). After a number of panning rounds, the resulting phages have competed against each other for antigen binding and their DNA can be isolated to reveal which amino acid sequences confer the ability of the antibodies to bind the chosen antigen. In phage display experiments, an antigen can be almost any macromolecule.
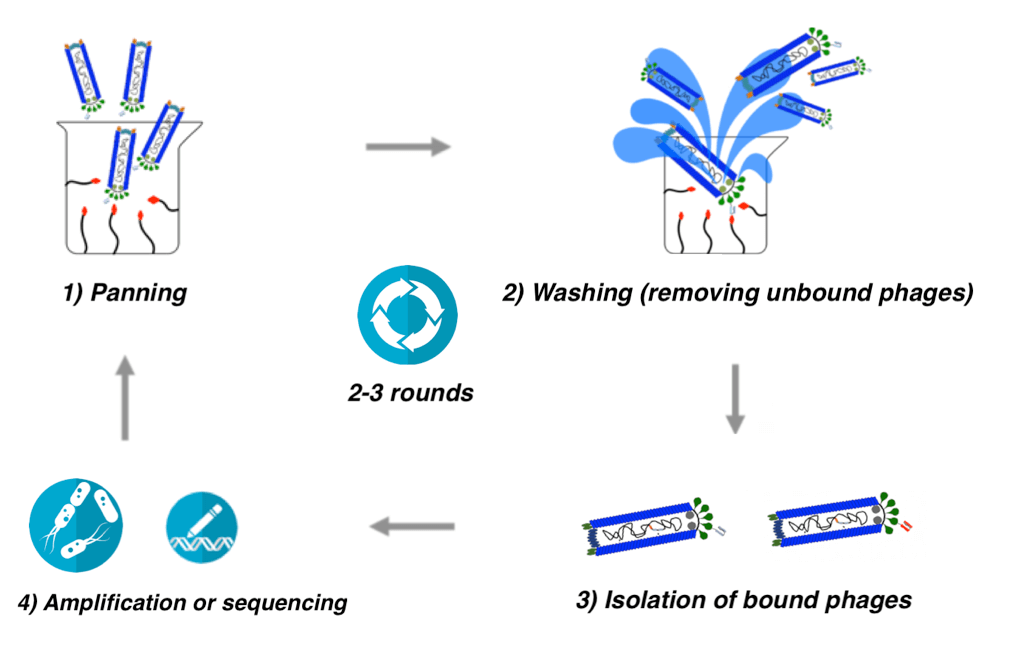
Figure 5: Schematic representation of a phage display experiment. 1) Antigens are immobilized in a vial, and the antibody-displaying phages are panned into the vial. 2) Phages that cannot recognize the antigens are washed away. 3) The antigen-binding phages are eluted. 4) The antigen-recognizing phages are amplified in E. coli. The process can then be repeated (typically 1-3 times), or the phage DNA can be sequenced to reveal the amino acid sequence of the displayed antibodies. (Illustration: Albert Fuglsang-Madsen).
A critical factor for discovering antibodies with good neutralization ability is to have a large, diverse collection of antibody fragments of high-quality (ability to bind multiple targets with high affinity and specificity). A way to construct such a collection is to isolate B-cells from human donors, isolate their antibody-encoding genes and clone these genes into bacteriophages (see Figure 6). This is typically done for several human donors to achieve a large and diverse antibody phage display library.
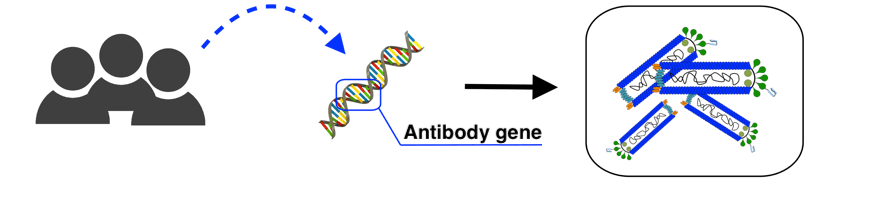
Figure 6: From human donors, the genes encoding the antigen binding regions of IgGs are isolated from B-cells and cloned into phages, where the gene is inserted into the phage genome, and the corresponding antibody fragment displayed on the surface of the phage virus particle. (Illustration: Albert Fuglsang-Madsen).
Another approach of assembling an antibody phage display library would be to use the B-cells of immunized animals. For instance, if one wishes to discover antibodies that effectively neutralize scorpion venom, using the variable regions of B-cells from mice, that have been immunized with scorpion venom, dramatically increases the chances of discovering strongly-neutralizing antibody fragments. However, this latter approach produces variable regions of non-human origin, which may evoke adverse reactions in human subjects, when entering clinical trials.
After antibody fragments have been discovered in a phage display experiment, they are typically converted into other antibody formats (such as the naturally occurring IgG) before they are taken further into preclinical and clinical development.
Although phage display holds great promise as an in vitro antibody discovery platform, there is room for improvement: In the future, it is possible that a combination of transgenic animals and phage display will be employed in the drug development process of IgG-based medicines. Transgenic mice with the human antibody gene repertoire could be immunized with a chosen antigen and their B-cells isolated and used for assembly of an optimized antibody phage display library that contains antibody fragments with improved affinity towards the antigen (see Figure 6). This way, “elite binders” (the best antigen-binding antibody fragments) can more easily be isolated from the library. This corresponds to selecting the best football players for the national team by scouting amongst the best football clubs, instead of randomly knocking on people’s houses and hoping to find elite players.
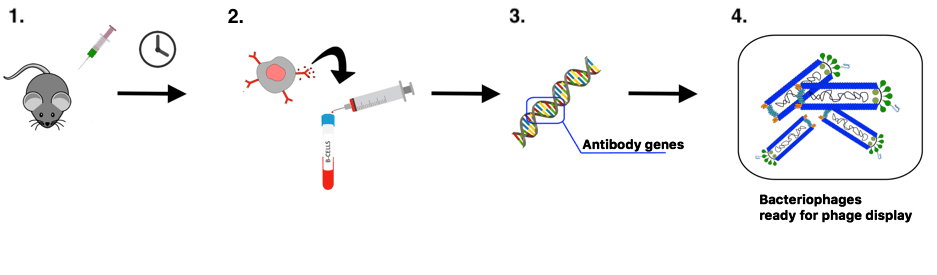
Figure 6: Illustration of how transgenic mice could be combined with phage display to obtain optimized antibody phage display libraries. 1) The transgenic mice are immunized with a relevant antigen. 2) Their B-cells are harvested. 3) The human parts of their antibody genes are isolated. 4) The antibody genes are cloned into bacteriophages, yielding an ‘affinity-matured’ antibody phage display library. This may increase the chances of discovering phages displaying antibodies with high affinity towards the antigen in subsequent phage display experiments. (Illustration: Albert Fuglsang-Madsen).
Despite our relatively recent understanding of the biochemical and mechanistic intricacies that surround antibodies, they have quickly become a major tool in our battle against diseases. In fact, the ability of antibodies to bind an enormous range of drug targets with great specificity make them ideal therapeutic molecules for treating diseases. Since the introduction of hybridoma technology, phage display, and transgenic animal models, therapeutic monoclonal antibody discovery has taken up speed dramatically and antibodies now represent the single largest class of molecules currently undergoing clinical trials. This will likely lead to more and better therapies against diseases and conditions that were previously untreatable.
By Albert Fuglsang-Madsen, Timothy P. Jenkins & Andreas Hougaard Laustsen

